Pheochromocytoma Treatment Decision Aid
Treatment Options Assessment
This tool helps determine the most appropriate treatment options for pheochromocytoma based on genetic mutations, tumor characteristics, and metastasis status. The information is based on current medical guidelines.
Recommended Treatment Options
If you’ve heard the word pheochromocytoma recently, you’re probably seeing headlines about new research.
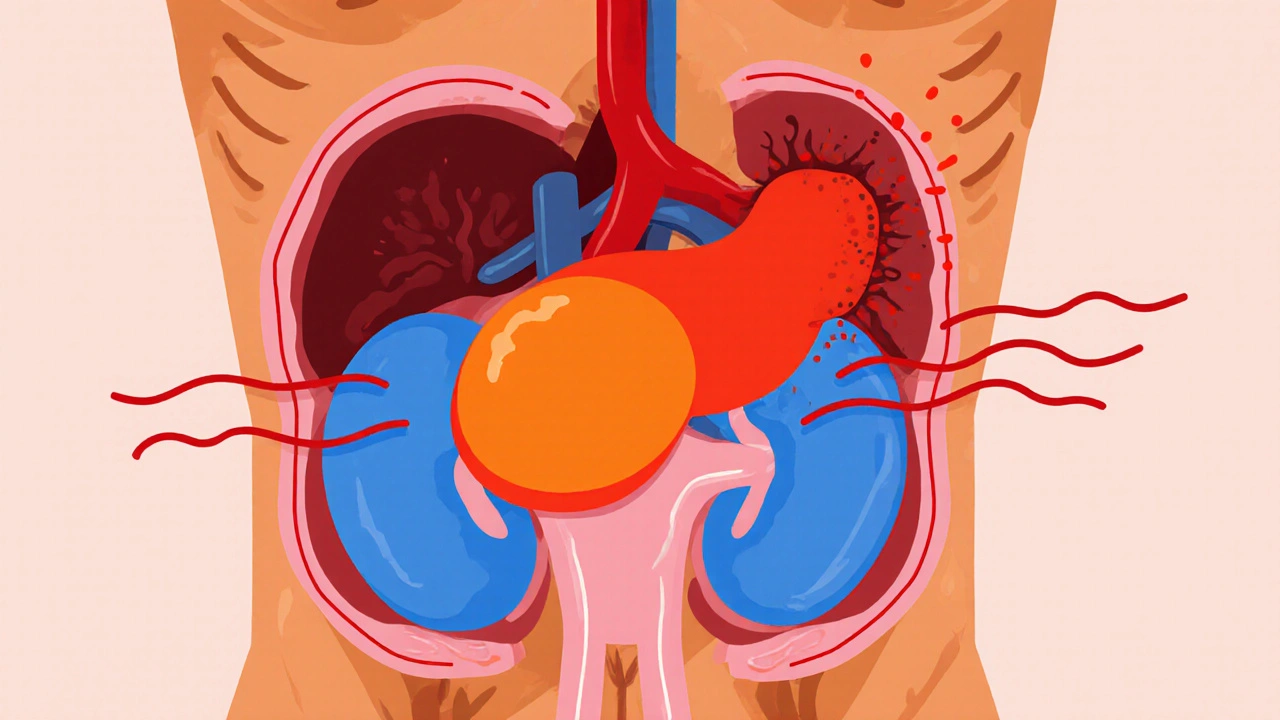
Pheochromocytoma is a rare tumor that arises from the chromaffin cells of the adrenal medulla, causing excess production of catecholamines such as adrenaline and noradrenaline. While it accounts for less than 0.2% of all hypertension cases, its unpredictable nature makes early detection critical.
What Exactly Is Pheochromocytoma?
The adrenal medulla sits atop each kidney and releases hormones that control the fight‑or‑flight response. When a pheochromocytoma develops, it hijacks this system, flooding the bloodstream with catecholamines. Common symptoms include pounding headaches, sweating, rapid heartbeat, and sudden spikes in blood pressure. Because these signs overlap with anxiety or panic attacks, many patients go undiagnosed for months.
Diagnosing the condition usually starts with a biochemical screen: measuring plasma free metanephrines or 24‑hour urinary fractionated metanephrines. Levels that are three times the upper reference limit are highly predictive of a tumor.
Why New Research Matters Now
Until the early 2000s, treatment was almost exclusively surgical. Over the past five years, a wave of genomic sequencing projects has uncovered a slew of hereditary mutations-most notably in the SDHB, SDHD, VHL, and RET genes. These discoveries have reshaped how clinicians stratify risk, plan surgery, and monitor long‑term outcomes.
For example, a 2024 multi‑center study of 1,200 patients showed that carriers of SDHB mutations faced a 30% higher chance of metastatic disease compared with sporadic cases. Knowing a patient’s genetic status now guides both the choice of initial therapy and the intensity of follow‑up imaging.
Genetic Insights Changing the Landscape
Genetic testing is no longer a specialist‑only service. Many major hospitals now offer a panel that includes the ten most common pheochromocytoma‑paraganglioma (PPGL) genes. The panel costs roughly $800 USD and returns results within two weeks.
Key takeaways from recent genetics research:
- SDHB mutations: linked to aggressive, often metastatic tumors; recommend early systemic therapy if surgery isn’t feasible.
- VHL mutations: usually produce smaller, less aggressive tumors; patients respond well to minimally invasive surgery.
- RET mutations: often associated with multiple endocrine neoplasia type 2 (MEN2); require lifelong surveillance for thyroid and parathyroid disease.
Beyond DNA sequencing, liquid biopsy-detecting circulating tumor DNA (ctDNA) in blood-has emerged as a non‑invasive way to monitor disease recurrence. A 2025 pilot reported 85% sensitivity for detecting metastatic spread months before imaging could.
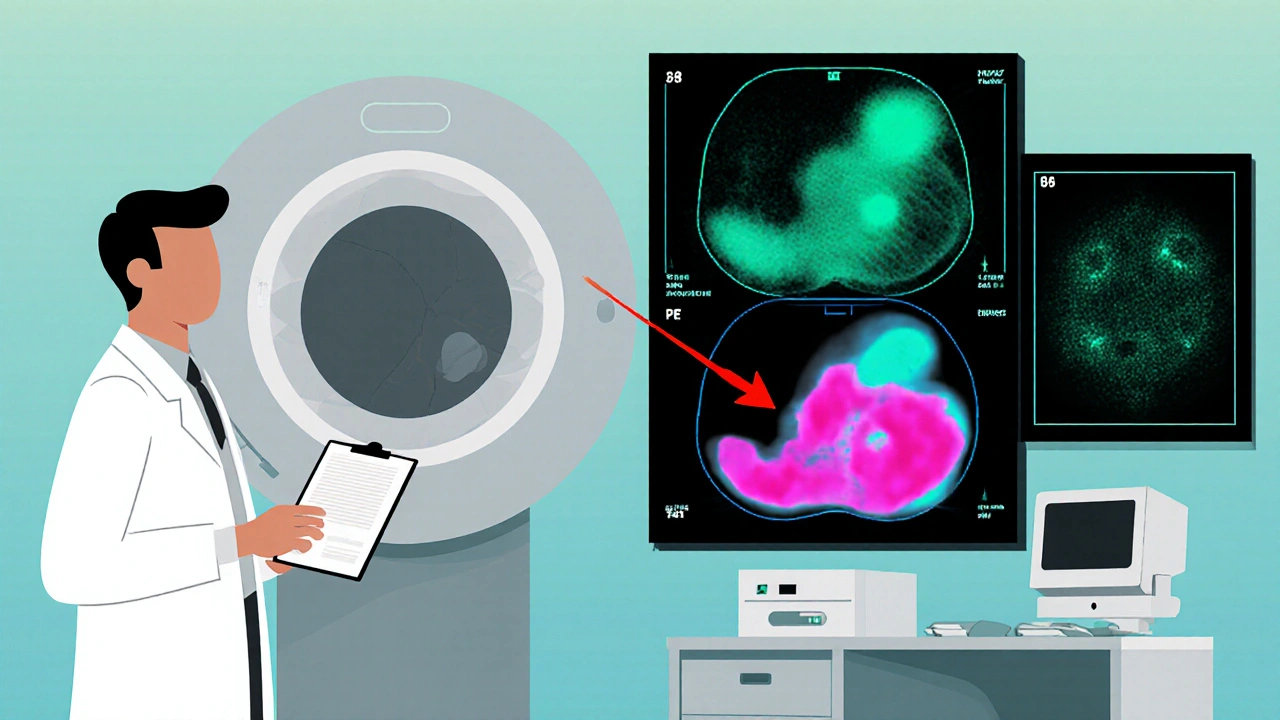
Modern Diagnostic Toolkit
Imaging has evolved from plain CT scans to functional modalities that visualize catecholamine activity. The current hierarchy looks like this:
- CT or MRI: first‑line to locate the tumor and assess size.
- 123I‑MIBG scintigraphy: highlights tumors that take up metaiodobenzylguanidine, useful for planning radionuclide therapy.
- 68Ga‑DOTATATE PET/CT: excellent for detecting somatostatin‑receptor positive lesions, especially in SDHB carriers.
- 18F‑FDG PET: picks up highly metabolic tumors, often metastatic.
Choosing the right scan depends on the suspected genetic background and whether the tumor is suspected to be metastatic.
Treatment Options: From Surgery to Targeted Radiotherapy
Historically, the gold standard has been laparoscopic adrenalectomy. Today, surgeons can choose between open, laparoscopic, or robot‑assisted procedures based on tumor size and location. Pre‑operative preparation remains crucial: patients receive an alpha‑adrenergic blocker (usually phenoxybenzamine) for 10‑14 days to blunt the catecholamine surge.
When surgery isn’t an option-due to metastasis, patient comorbidities, or unresectable disease-systemic therapies step in. Below is a quick comparison of the main non‑surgical options:
| Treatment | Mechanism | Approx. Success Rate | Common Side Effects | Typical Use Case |
|---|---|---|---|---|
| Laparoscopic adrenalectomy | Physical removal of tumor | 80‑95% cure for localized disease | Bleeding, adrenal insufficiency | Localized, resectable tumors |
| 131I‑MIBG therapy | Radioactive iodine‑labeled MIBG taken up by catecholamine‑producing cells | 40‑60% disease control | Bone marrow suppression, nausea | Metastatic or inoperable disease that is MIBG‑avid |
| PRRT (177Lu‑DOTATATE) | Radiolabeled somatostatin analog binds to tumor receptors | 55‑70% partial response | Renal toxicity, mild fatigue | SDHB‑related tumors expressing somatostatin receptors |
| Tyrosine kinase inhibitors (TKIs) | Blocks signaling pathways that drive tumor growth | 30‑45% disease stabilization | Hypertension, hand‑foot syndrome | Progressive disease after radionuclide therapy |
Each option has its own pros and cons, and the best plan usually blends more than one modality. For instance, a patient might undergo surgery to remove the primary mass, followed by PRRT to mop up residual metastatic lesions.
Emerging Therapies on the Horizon
Research labs are racing to translate molecular insights into targeted drugs. Three promising directions stand out:
- CRISPR‑based gene editing: Early‑stage trials aim to correct pathogenic SDHB mutations in tumor cells, potentially halting progression.
- Immune checkpoint inhibitors: Small phase‑II studies using pembrolizumab have shown durable responses in a subset of patients with high tumor mutational burden.
- Nanoparticle‑delivered chemotherapeutics: Liposomal doxorubicin combined with a catecholamine‑targeting ligand is being tested to deliver chemotherapy directly to tumor tissue, reducing systemic toxicity.
While these therapies are not yet standard care, they illustrate how the field is moving from blunt surgical removal to precision medicine.
Managing Side Effects and Long‑Term Follow‑Up
Even after a successful operation, patients remain at risk for recurrence-especially those with hereditary mutations. Current guidelines recommend:
- Annual plasma metanephrine testing for the first five years, then every two years.
- Imaging every 2‑3 years, using the most sensitive modality for the patient’s genetic profile.
- Genetic counseling for first‑degree relatives, because up to 40% of cases are familial.
Blood pressure control often normalizes after tumor removal, but some patients develop persistent hypertension due to vascular remodeling. In such cases, a low‑dose ACE inhibitor combined with lifestyle changes usually suffices.
Key Takeaways
- Pheochromocytoma is a rare but treatable adrenal tumor that causes dangerous spikes in catecholamines.
- Genetic testing now guides risk assessment, treatment selection, and surveillance strategies.
- Surgery remains first‑line for localized disease, but radionuclide therapies like 131I‑MIBG and PRRT provide effective options for metastatic cases.
- Emerging treatments-including CRISPR editing and immunotherapy-are under active investigation and may become mainstream within the next decade.
- Lifelong monitoring is essential, particularly for patients with SDHB or other high‑risk mutations.
What are the most common symptoms of pheochromocytoma?
Typical signs include pounding headaches, excessive sweating, rapid heartbeat, anxiety‑like feelings, and sudden high‑blood‑pressure spikes. Episodes often come in waves and can be triggered by stress, standing up quickly, or certain foods.
How is pheochromocytoma diagnosed?
Diagnosis starts with measuring plasma free metanephrines or a 24‑hour urine collection for fractionated metanephrines. Positive biochemical tests are followed by imaging-CT, MRI, or functional scans like 123I‑MIBG, 68Ga‑DOTATATE PET, and 18F‑FDG PET-to locate the tumor and check for spread.
When is surgery recommended?
Surgery is first‑line for tumors that are confined to the adrenal gland and are resectable. Minimally invasive laparoscopic or robot‑assisted adrenalectomy is preferred for most patients, provided the tumor is less than 6‑8 cm and there’s no invasion into surrounding structures.
What non‑surgical treatments are available for metastatic disease?
Options include 131I‑MIBG radiotherapy, peptide receptor radionuclide therapy (PRRT) with 177Lu‑DOTATATE, tyrosine‑kinase inhibitors, and enrollment in clinical trials for immunotherapy or gene‑editing approaches. Choice depends on tumor genetics, uptake on functional imaging, and patient health.
How often should patients be monitored after treatment?
Guidelines suggest annual plasma metanephrine tests for the first five years, then every two years. Imaging-usually MRI or the most sensitive PET scan for the individual’s mutation profile-is recommended every 2‑3 years, or sooner if symptoms recur.
Penny Reeves
It is astonishing how the recent deluge of pheochromocytoma literature seems to masquerade as breakthrough when, in fact, it merely rehashes established dogma; the biochemical triad of metanephrines, imaging, and surgery has been the cornerstone for decades, not some novel esoteric revelation. The authors of this post apparently missed an opportunity to interrogate the socioeconomic implications of a $800 genetic panel, a cost prohibitive for many patients, thereby perpetuating inequity. Moreover, the emphasis on functional imaging modalities such as 68Ga‑DOTATATE PET, while technologically impressive, obscures the fact that conventional CT remains an adequate first‑line tool in most cases. One must also note the glossed‑over toxicity profiles of radionuclide therapies; bone marrow suppression is not a trivial side effect and demands rigorous hematologic monitoring. The cited 2024 multi‑center study, although robust, failed to stratify outcomes by ethnicity, a glaring omission given the variable prevalence of SDHB mutations across populations. Furthermore, the discussion of CRISPR‑based gene editing skims past ethical considerations surrounding germline modifications. While the article celebrates the advent of liquid biopsy, it neglects the false‑positive rates inherent in circulating tumor DNA assays, potentially leading to overtreatment. The claim that lifelong monitoring is essential could be softened with a nuanced risk‑based algorithm rather than a blunt blanket recommendation. In the grand scheme, the piece reads like a press release from a biotech vendor rather than a balanced clinical overview. Finally, the lack of patient‑centered perspectives renders the narrative sterile, missing the lived experience of those grappling with episodic hypertensive crises. In sum, the post, though well‑intentioned, suffers from an oversimplified optimism that belies the complexity of pheochromocytoma management.
Sunil Yathakula
Hey folks, this is a lot to take in but dont worry you can handle it! The new gene panels are actually pretty cool – they give you answers faster and you can plan treatment early. If you feel scared about the numbers, just remember the doctors will guide you step by step. And yeah, the side effects sound scary but most people manage them well with good care. Stay positive and keep checking in with your team. You got this!
Catherine Viola
One must, with due diligence, scrutinize the purported advances delineated herein, for the specter of corporate influence looms ever larger over contemporary oncologic research. It is a matter of grave concern that the financial incentives of diagnostic conglomerates may subtly dictate which genetic panels receive preferential endorsement, thereby shaping clinical practice to serve profit motives rather than patient welfare. Moreover, the cavalier mention of CRISPR technology, absent a rigorous discourse on off‑target effects, betrays a naïveté befitting a pamphlet rather than a scholarly treatise. The authors neglect to acknowledge the historical pattern whereby novel therapeutics are prematurely heralded, only to be later reined in by unforeseen toxicities. In the realm of radionuclide therapy, the paucity of long‑term surveillance data should incite caution among practitioners. I would also posit that the emphasis on high‑cost imaging modalities may exacerbate disparities, especially in under‑funded health systems where resources are finite. Hence, while the veneer of progress is alluring, it must be pierced with a critical eye to expose the underlying machinations that may compromise the sanctity of patient‑centered care.
sravya rudraraju
Friends, let us celebrate the strides made in pheochromocytoma management while acknowledging the collaborative spirit that fuels such advancements. The integration of genetic testing into routine work‑up reflects a paradigm shift toward precision medicine, empowering clinicians to tailor interventions based on molecular profiles. It is heartening to observe the expanding repertoire of functional imaging, which not only refines staging but also informs therapeutic selection, particularly for SDHB‑associated tumors. Importantly, the multidisciplinary approach-encompassing endocrinologists, surgeons, nuclear medicine specialists, and genetic counselors-highlights the value of teamwork in navigating complex cases. While we commend the emergence of promising modalities like PRRT and CRISPR‑based strategies, we must remain vigilant regarding safety and ethical considerations. Ongoing trials will undoubtedly clarify the optimal sequencing of these therapies, fostering an evidence‑based framework for patient care. Let us also remember the significance of patient education, ensuring individuals understand their genetic risk and the implications for family members. In sum, the momentum generated by recent research heralds a brighter future for those affected by this rare tumor, provided we uphold the principles of transparency, equity, and compassionate care.
Ben Bathgate
Okay, the hype about fancy scans and gene panels is overblown – most of the time a simple CT does the job and saves cash.
Ankitpgujjar Poswal
Listen up! If you’re not jumping on the PRRT train now, you’re falling behind. Get your oncologist on the phone and demand the best options – no excuses!
Bobby Marie
Just get tested.
Kevin Sheehan
Consider the existential dimensions of a disease that hijacks the body’s very stress response. When catecholamines surge uncontrollably, the individual confronts a literal battle between physiological survival mechanisms and pathological overdrive. This tension invites reflection on the broader human condition: how much of our anxiety is rooted in biology versus perception? The advent of genetic insight offers a mirror to our inherent vulnerabilities, urging us to seek balance not merely through surgery, but through holistic stewardship of mind and body. In practice, this translates to integrating lifestyle interventions-mindfulness, moderated caffeine intake, and regular cardiovascular monitoring-alongside cutting‑edge therapeutics. Thus, the modern management of pheochromocytoma becomes a microcosm of the pursuit of harmony between technology and innate human resilience.
Jay Kay
Wow – another “breakthrough” that probably won’t change the day‑to‑day grind for most patients. Cue the dramatic music.
Monika Bozkurt
From a clinical standpoint, the incorporation of somatostatin receptor imaging (e.g., 68Ga‑DOTATATE PET) synergizes with molecular diagnostics to refine risk stratification algorithms. By leveraging lesion‑to‑background ratios and standardized uptake values, clinicians can quantitatively assess receptor density, thereby informing eligibility for peptide receptor radionuclide therapy (PRRT). This evidence‑based approach optimizes therapeutic index while minimizing off‑target radiation exposure. Additionally, longitudinal metanephrine surveillance, calibrated against genotype‑specific baseline thresholds, enhances early detection of recurrence, ultimately improving progression‑free survival metrics. It is imperative that multidisciplinary tumor boards synthesize these data streams to construct individualized care pathways.
Latasha Becker
Contrary to popular belief, the proliferation of $800 genetic panels is not driven by patient benefit but by a concerted effort from biotech investors to monetize genome‑wide screenings. The literature conveniently omits discussion of the false‑positive rates that inflate downstream imaging and procedural costs. One must question whether the enthusiasm for liquid biopsy is a marketing ploy rather than a robust clinical tool.
parth gajjar
the drama of a hormone storm it's like a fireworks show inside your veins but without the fun and the fireworks are your heart beating like a drum i can feel the panic rising the world blurs and it's all so intense and terrifying
DHARMENDER BHATHAVAR
Genetic profiling guides therapy selection, enabling clinicians to prioritize PRRT for somatostatin‑receptor positive cases.
Christian Georg
Hey everyone 😊! Just a quick heads‑up: if you’re planning an adrenalectomy, make sure you’re on an alpha‑blocker for at least 10‑14 days – it really tames those catecholamine spikes. Also, keep an eye on post‑op blood pressure; some patients need a low‑dose ACE inhibitor for lingering hypertension. Feel free to DM me if you want more detailed protocols or paper links! 👍
Christopher Burczyk
The article presents a superficial overview, failing to critically appraise the limitations of current therapeutic modalities and the socioeconomic barriers to accessing advanced diagnostics.